Molecular Dynamics study of hydrogen adsorption in microporous carbonaceous materials and the effect of oxygen functional groups
This work investigates the adsorption of hydrogen into microporous carbonaceous adsorbents via the use of Molecular Dynamics simulations at atomic scale. Different 3D models of known carbonaceous structures were built with HyperChem computational chemistry package, and the adsorption process was investigated at nano sec time scale. The effects of different pore sizes, shapes and temperatures were revealed. Minimum pore sizes for adsorption initiation was identified at 5.5A. This results also suggests manufacturing details for separation membranes and hydrogen storage media based on physisorption.
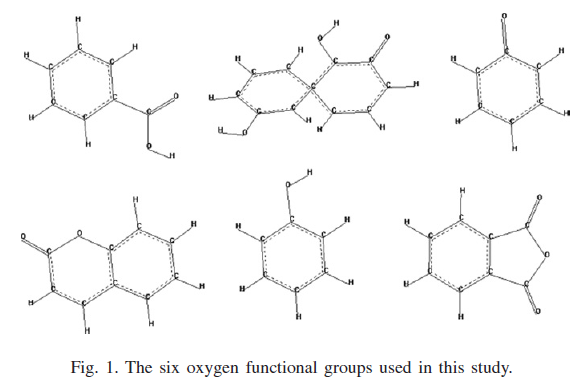
Different oxygen functional groups were investigated regarding their potential effect on hydrogen adsorpion on these model solids. These groups were incorporated into the previously built models, and the new models were re- optimized via a geometry optimization procedure which resulted to local alteration of initially flat graphene sheets. Different amount of oxygen content were used in the new models creation, trying to evaluate a relationship between content and adsorption change. Although adsorption density into pure and oxygen doped solids was higher than that of liquid hydrogen's, the DOE limit was not reached, and the pure carbonaceous micropores exhibited higher gravimetric adsorption of hydrogen.

To download the PDF click here